КАТЕГОРИИ:
Архитектура-(3434)Астрономия-(809)Биология-(7483)Биотехнологии-(1457)Военное дело-(14632)Высокие технологии-(1363)География-(913)Геология-(1438)Государство-(451)Демография-(1065)Дом-(47672)Журналистика и СМИ-(912)Изобретательство-(14524)Иностранные языки-(4268)Информатика-(17799)Искусство-(1338)История-(13644)Компьютеры-(11121)Косметика-(55)Кулинария-(373)Культура-(8427)Лингвистика-(374)Литература-(1642)Маркетинг-(23702)Математика-(16968)Машиностроение-(1700)Медицина-(12668)Менеджмент-(24684)Механика-(15423)Науковедение-(506)Образование-(11852)Охрана труда-(3308)Педагогика-(5571)Полиграфия-(1312)Политика-(7869)Право-(5454)Приборостроение-(1369)Программирование-(2801)Производство-(97182)Промышленность-(8706)Психология-(18388)Религия-(3217)Связь-(10668)Сельское хозяйство-(299)Социология-(6455)Спорт-(42831)Строительство-(4793)Торговля-(5050)Транспорт-(2929)Туризм-(1568)Физика-(3942)Философия-(17015)Финансы-(26596)Химия-(22929)Экология-(12095)Экономика-(9961)Электроника-(8441)Электротехника-(4623)Энергетика-(12629)Юриспруденция-(1492)Ядерная техника-(1748)
Нуклеофильный катализ
|
|
|
|
Наиболее распространенными нуклеофильными катализаторами являются ионы галогенов I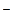 , Вrи F, оксианионы AlkO, АrО, НСО
, Вrи F, оксианионы AlkO, АrО, НСО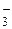 , СО
, СО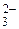 , РО
, РО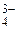 и НО, амины R3N, R2NH, RNH2, C5H5N, а также некоторые другие анионы: No
и НО, амины R3N, R2NH, RNH2, C5H5N, а также некоторые другие анионы: No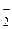 , CN, С1О, НОО.
, CN, С1О, НОО.
По конечному результату нуклеофильные каталитические реакции можно подразделить на реакции присоединения и замещения.
В реакциях нуклеофильного присоединения YH по кратным связям
X=Z + YH ® Y–X–Z–H (5.2)
роль нуклеофильного катализатора часто выполняет анион Y- сопряженное основание реагента, который обладает значительно более сильными нуклеофильными свойствами, чем YH, и легко присоединяется по кратной связи:
Y+ X=Z 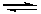 Y–X–Z (5.3)
Y–X–Z (5.3)
Образующийся промежуточный анион регенерирует затем катализатор по протолитической реакции с YH:
Y–X–Z+ YH ® Y–X–Z–H + Y (5.4)
По такой схеме происходит нуклеофильное присоединение по карбонильной группе реагента с кислотными свойствами, например синильной кислоты:
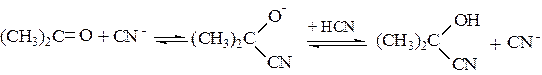 (5.5)
(5.5)
При этом нередко за счет водородной связи вначале образуются промежуточные комплексы между реагентами, что снижает электронную плотность на реакционном центре и ускоряет реакцию. Например, так протекают реакции с аналогами карбонильных соединений – нитрилами:
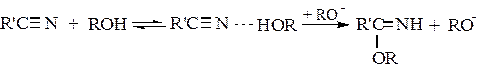 (5.6)
(5.6)
В других случаях реакция может идти сразу по обоим механизмам.
Нуклеофильный катализ эффективен в аналогичных реакциях присоединения YH к олефинам с электроноакцепторным заместителем при кратной связи (CH2=CHZ, где Z – COOR, NO2, CF3, COR, F или CN) и к ацетиленам. В этих реакциях кратная связь активируется образованием π-комплекса YH с реагентом:
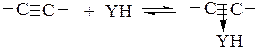 (5.7)
(5.7)
На следующей стадии происходит присоединение нуклеофильного катализатора Y- с одновременной его регенерацией из молекулы YH:
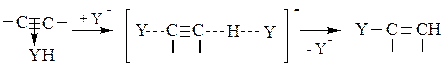 (5.8)
(5.8)
Парой реагент (YH) – катализатор (Y-) в этих реакциях могут быть ROH–RO, Н2О–НО, H2S–HS, ArOH–ArOи т. п. Например:
H2O + CH2=CHCN  HOCH2CH2CN
HOCH2CH2CN
CH3OH + HCºCH 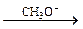 CH3OCH=CH2 (5.9)
CH3OCH=CH2 (5.9)
Нуклеофильный катализ сопряженными основаниями характерен также для реакций присоединения YH к гетероциклическим соединениям с малыми циклами, Как и в реакциях присоединения по кратным связям, исходный реагент активируется засчет образования водородной связи с YH:
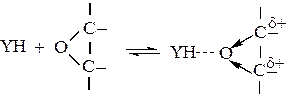 (5.10)
(5.10)
Образовавшийся комплекс взаимодействует затем с катализатором Yи присоединяет протон от YH с одновременной регенерацией Y:
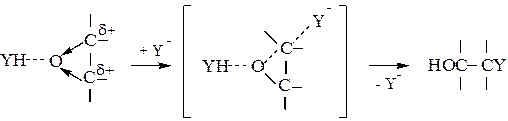 (5.11)
(5.11)
Катализ сопряженными основаниями наиболее эффективен для реакций присоединения спиртов, карбоновых кислот, фенолов, меркаптанов, сероводорода и синильной кислоты к α-оксидам:
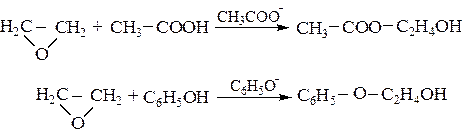 (5.12)
(5.12)
Замещение RZ + Y → RY+Z при нуклеофильном катализе в простейшем случае представляет собой чередование двух реакций нуклеофильного замещения:
RZ + Nu ® RNu + Z RNu + Y ® RY + Nu (5.13)
В реакциях замещения, протекающих при насыщенном атоме углерода, свойствами активного нуклеофила и одновременно легко замещаемой группы обладают анионы йода и брома. Примером могут служить реакции гидролиза хлор- или бромпроизводных при катализе анионом иода:
 RX + I® RI + X RI + 2H2O ROH ® H3O+ + I (5.14)
RX + I® RI + X RI + 2H2O ROH ® H3O+ + I (5.14)
Широко распространен нуклеофильный катализ в реакциях замещения по карбоксильной группе производных карбоновых кислот. Для превращений ангидридов и хлорангидридов активными нуклеофильными катализаторами являются третичные амины. Механизм этих реакций заключается в промежуточном образовании катиона ацилпиридиния (или ациламмония RCON + R3) – одного из самых эффективных ацилирующих агентов благодаря сильным электроноакцепторным свойствам четвертичного азота и отсутствию сопряжения с С=О -группой:
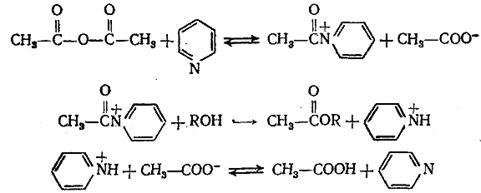 (5.15)
(5.15)
В реакциях алкоголиза сложных эфиров возможен нуклеофильный катализ другого типа, в котором в качестве катализатора используется сопряженное основание реагента. Так же как в реакциях присоединения, повышенная нуклеофильность сопряженного основания по сравнению с реагентом позволяет резко ускорить процесс (по сравнению с некаталитическим):
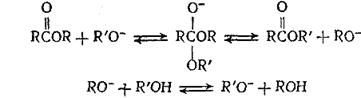 (5.16)
(5.16)
Кинетика реакций нуклеофильного катализа. Практически все рассмотренные реакции можно представить двумя общими схемами. Одна из них включает образование комплекса между реагентами (за счет водородной связи), его реакцию с нуклеофильным катализатором Yи образование продукта Р:
RZ + YH 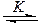 YH×××ZR
YH×××ZR 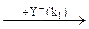 Y+ P (5.17)
Y+ P (5.17)
При другом механизме нуклеофильного катализа промежуточный комплекс RNu образуется при взаимодействии катализатора Nu с одним из реагентов RZ, а образование продукта и регенерация катализатора происходят в результате последующих превращений комплекса:
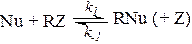
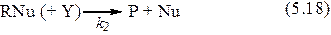
Схема упрощается, если первая стадия является реакцией присоединения (отсутствует Z), а вторая стадия мономолекулярна (отсутствует Y). Кроме того, пер-
вая стадия может быть необратимой. Схема характерна не только для нуклеофильного катализа. По этой причине приведенный ниже кинетический анализ и методология кинетического исследования имеют общий характер для всех гомогенно-каталитических реакций.
Если допустить, что концентрация катализатора мала по сравнению с концентрацией реагентов, и последние не образуют друг с другом каких-либо комплексов, то в общем случае (когда первая стадия – обратимая реакция замещения, а вторая стадия бимолекулярна) уравнение для определения скорости реакции имеет вид:
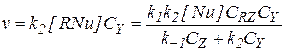 (5.19)
(5.19)
Последнее уравнение требует дополнительного уточнения, учитывающего присутствие катализатора в двух формах (Nu и RNu).
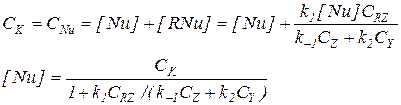 (5.20)
(5.20)
Окончательное выражение для скорости реакции:
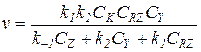 (5.21)
(5.21)
Обычно бывает так, что скорость одной или двух элементарных стадий пренебрежимо мала по сравнению с остальными, и одним или двумя слагаемыми знаменателя в последнем уравнении можно пренебречь. Тогда оно приобретает более простые формы, каждая из которых имеет свои особенности. Всего возможны семь комбинаций слагаемых знаменателя в этом уравнении.
1) Если k 2CY >> k -1CZ и k 2CY >> k 1CRZ, то в знаменателе остается только один член k2CY, который сокращается с числителем, и в результате для скорости получается уравнение:
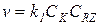 (5.22)
(5.22)
В этих условиях практически весь катализатор находится в свободной форме, скорость лимитируется первой стадией и реакция имеет нулевые порядки по Z и Y.
2) Когда k 1CRZ >> k -1CZ и k 1CRZ >> k 2CY, получаем:
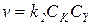 (5.23)
(5.23)
В подобных условиях практически весь катализатор находится в форме каталитического комплекса, скорость лимитируется второй стадией, и на нее не влияют концентрации Z и RZ.
3) При k -1CZ >> k 1CRZ и k -1CZ >> k 2CY:
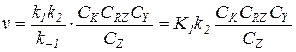 (5.24)
(5.24)
Здесь на первой стадии устанавливается равновесие с низкой концентрацией промежуточного комплекса, которая составит:
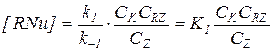 (5.25)
(5.25)
Скорость лимитируется второй стадией, реакция имеет первые порядки по обоим реагентам и тормозится продуктом.
4) Когда слагаемое знаменателя k-1CZ значительно меньше двух остальных, получаем уравнение:
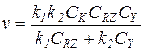 (5.26)
(5.26)
В этом случае первая стадия практически необратима, а стационарные концентрации двух форм катализатора [RNu] и [Nu] можно найти из условия равенства скоростей первой и второй стадий:
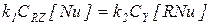 или
или 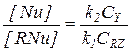 (5.27)
(5.27)
5) В знаменателе уравнения (5.21) можно пренебречь членом k1CRZ, а остальные слагаемые соизмеримы (k-1CZ» k2CY):
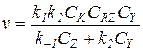 (5.28)
(5.28)
Уравнение (5.28) соответствует медленной реакции катализатора в исходной форме Nu с реагентом RZ и быстрому последующему взаимодействию промежуточного продукта RNu с Z и Y с образованием исходного или конечного продукта. При таком соотношении скоростей реакция тормозится продуктом Z, а катализатор находится преимущественно в исходной форме (Ск» [Nu]).
6) Наименьшее значение в знаменателе уравнения (5.21) принимает слагаемое k2CY, а два других соизмеримы:
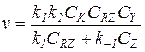 (5.29)
(5.29)
Такое уравнение справедливо при лимитировании скорости реакции взаимодействием промежуточного продукта RNu и Y на второй стадии. При этом условии на первой стадии устанавливается равновесие с близкими значениями концентраций [Nu] и [RNu]:
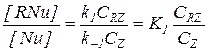 (5.30)
(5.30)
и реакция тормозится продуктом Z.
7) Скорости всех трех элементарных реакций в рассматриваемой схеме соизмеримы, а скорость суммарной реакции описывается уравнением (5.21) в наиболее общей форме. При этом равновесие на первой стадии не устанавливается, а соотношение стационарных концентраций [Nu] и [RNu] описывается уравнением (5.21).
Кинетические закономерности реакции (5.17), протекающей через предварительное образование комплекса YH---ZR за счет водородной связи, не столь разнообразны, поскольку равновесие образования таких комплексов устанавливается всегда быстро. Это позволяет выразить концентрацию комплекса через концентрацию свободных форм реагентов и константу равновесия:
[YH---ZR] = K[YH][RZ] (5.31)
и представить кинетическое уравнение реакции в таком виде:
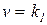 [YH---ZR]
[YH---ZR] (5.32)
(5.32)
Поскольку катализатор в этой схеме не образует никаких комплексов, общая его концентрация Ск равна концентрации Y-, a для выражения концентраций свободных форм реагентов через их суммарные или аналитические концентрации необходимо учесть комплексообразование между ними:
CRZ = [RZ] + [YH---ZR] = [RZ] + K[YH][RZ]
CYH = [YH] + [YH---ZR] = [YH] + K[YH][RZ] (5.33)
Совместное решение уравнений скорости и баланса дает общее выражение для скорости реакции, протекающей по схеме (5.17):
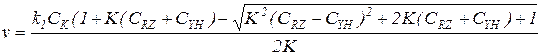 (5.34)
(5.34)
Кинетику гомогенно-каталитических реакций обычно исследуют в периодических условиях, причем объем реакционной смеси остается все время практически постоянным. С целью упрощения кинетических уравнений первые серии опытов часто проводят при одинаковой концентрации катализатора в большом избытке одного из реагентов. Концентрации реагентов постоянны, что позволяет определить значения эффективных констант. Так, при [YH]0>>[RZ]0 можно полагать, что CYH,0@[YH], и при выводе кинетического уравнения необходимо учитывать только баланс по RZ. Это значительно упрощает уравнение (5.34):
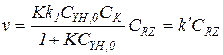 (5.35)
(5.35)
Уравнения (5.21) - (5.29) при условии избытка Y также преобразуются в одну из четырех более простых форм, которая легко интегрируется:
v = k’ v = k’CRZ v =  или
или 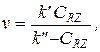
где k' — функция СК, CY,0 и элементарных констант, а k'' — функция CY,0, CRZ,0 и элементарных констант. Значения k' и k'' можно определить из графика, построенного по результатам эксперимента в координатах преобразованного в линейную форму кинетического уравнения. Зависимость k' и k'' от С К, C Y,0 или C RZ,0 проверяют в дополнительных сериях опытов с варьированием этих величин. В результате устанавливают кинетическое уравнение и механизм, соответствие которых опыту проверяют с учетом всех экспериментальных данных, и одновременно уточняют значения констант.
Для определения констант кинетических уравнений со знаменателями в ряде случаев более удобным оказывается дифференциальный метод. В частности, уравнение v = k'C/(k"+C) можно преобразовать в линейную форму тремя способами:
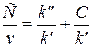
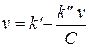
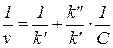
Наиболее распространенным является последний способ линеаризации (координаты Лайнуивера - Берка, 1/ v — 1/ С).
Исследование кинетики каталитической реакции может быть осложнено протеканием параллельной некаталитической реакции, влиянием образующегося продукта на скорость реакции и изменением природы катализатора во времени.
Ускорение реакции продуктом (автокатализ) наблюдается, в частности, в некоторых реакциях нуклеофильного присоединения меркаптанов:
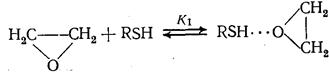
RZ YH YH∙∙∙ZR
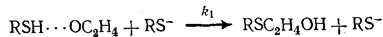
YH∙∙∙ZR Y - BH Y -
Образующийся спирт ВН активирует исходный этиленоксид RZ сильнее, чем меркаптан YH (K 2 >K 1 )
RZ + BH 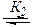 RZ---HB,
RZ---HB,
что приводит к параллельному протеканию более быстрой реакции образования того же продукта (k 2 >k 1 ):
BH---ZR + Y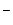
 2BH
2BH
При малых концентрациях комплексов BH∙∙∙ZR и YH∙∙∙ZR нет необходимости учитывать баланс по YH, RZ и ВН. Тогда суммарное уравнение основной и автокаталитической реакций имеет вид:
v = K1k1CYHCY – CRZ + CRZ + K2k2CBHCY – CRZ (5.36)
При избытке YH и постоянстве С Y это уравнение имеет вид:
r = (k' + k"C BH ) C RZ , (5.37)
где k' = К1 k1CY-CYH,0, k" = К 2 k 2 C Y-
Для определения его параметров можно воспользоваться дифференциальным методом.
Превращение катализатора в другие формы в ходе реакции часто не позволяет выразить скорость реакции одним уравнением. В этих случаях вводят дополнительные дифференциальные уравнения, связывающие концентрации разных форм катализатора с концентрациями реагентов.
Факторы, определяющие эффективность нуклеофильного катализа. Из приведенных ранее схем нуклеофильного катализа (5.17) и (5.18) следует, что его эффективность определяется стадией взаимодействия нуклеофила и субстрата или комплекса субстрата с YH. Скорость этой стадии зависит от нуклеофильности катализатора, которая, как и нуклеофильность реагентов, определяется их основностью и поляризуемостью реакционного центра.
Общим для всех реакций нуклеофильного катализа является влияние растворителя на активность нуклеофильного катализатора, в большинстве случаев представляющего собой ионную пару M+Y.
Максимальная скорость реакций с участием M+Yдостигается в полярных апротонных растворителях (диметилформамид, диметилсульфоксид, ацетонитрил, тетраметиленсульфон), в которых соль диссоциирует, а свободный анион Yпрактически не взаимодействует с растворителем.
При переходе к протонным растворителям той же полярности (вода, метанол, этанол) нуклеофильная активность свободного Yснижается на 2 - 5 порядков за счет сольватации с образованием водородных связей [Y∙∙∙(HOR)n]. При этом сильнее сольватируются ионы, меньшие по размеру и с большим зарядом.
 В растворителях умеренной полярности (высшие спирты, ацетон) равновесие диссоциации M+Y M+ + Yсдвинуто в сторону образования ионной пары. Ассоциация аниона Yс катионом уменьшает нуклеофильную активность аниона так же, как и сольватация за счет водородной связи. Особенно прочные ионные пары образуются ионами небольшого размера, поэтому увеличение размеров катиона всегда способствует росту нуклеофильности аниона, что следует иметь в виду при выборе катализатора.
В растворителях умеренной полярности (высшие спирты, ацетон) равновесие диссоциации M+Y M+ + Yсдвинуто в сторону образования ионной пары. Ассоциация аниона Yс катионом уменьшает нуклеофильную активность аниона так же, как и сольватация за счет водородной связи. Особенно прочные ионные пары образуются ионами небольшого размера, поэтому увеличение размеров катиона всегда способствует росту нуклеофильности аниона, что следует иметь в виду при выборе катализатора.
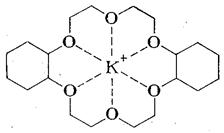
С этой целью, а также для придания растворимости анионам в малополярных растворителях, нуклеофильные катализаторы применяют в виде солей с объемистыми органическими катионами [(С2Н5)3С6Н5СН2N+, н -С6Н13(н -С4Н9)3Р+ и т. п.] или добавляют специальные комплексообразователи для неорганического катиона, например, циклические полиэфиры гликолей - краун-эфиры:
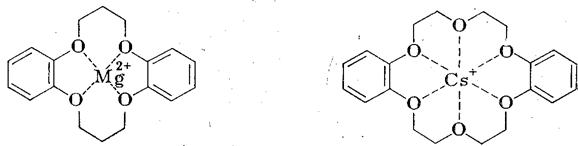
дибензо-16-краун-4* дибензо - 18 - краун – 6
(*4 — число гетероатомов, 16 – число атомов в цикле)
|
|
|
|
|
Дата добавления: 2014-01-07; Просмотров: 3462; Нарушение авторских прав?; Мы поможем в написании вашей работы!